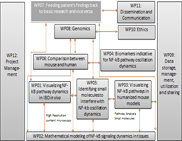
WP02 - Mathematical modelling of NF-kappa-B signalling dynamics in tissues
Objectives
Recent systems-biology research has shown that the temporal dynamics of NF-kappa-B signalling controls downstream cellular decisions.
In close and iterative collaboration with the experimental groups (WP 01, 03, 04 and 07) we will:
- Develop mechanistically-based mathematical models of NF-kappa-B signalling that rationalize cell type-specific signalling dynamics in the gut (tasks 1-3).
- Dissect how dynamics of NF-kappa-B signalling give rise to specific expression programs (task 4).
- Model paracrine signalling and cellular heterogeneity in shaping NF-kappa-B dynamics in tissues (task 3, 5).
Workpackage Description
Task 1: Development of a family of kinetic models for the NF-kappa-B pathway
Mathematical models of the canonical NF-kappa-B pathway vary with respect to complexity, biophysical realism and capability to explain experimental findings. Using existing data from UNIMAN on NF-kappa-B oscillations and signalling transients, we will develop a family of kinetic models for the canonical and non-canonical NF-kappa-B pathways. The models will initially be formulated as deterministic ordinary differential equations for concentration variables. The simplest model will centre on the common core of negative transcriptional feedback regulation of NF-kappa-B by I-kappa-B and describe reversible phosphorylation as well as nucleo-cytoplasmic shuttling of the pathway components with a small number of variables and by using simplified mass-action kinetics.
Task 2: Cell-type specific model parameterisation based on live-cell imaging experiments
Bayesian statistics-based model parameterisation and selection techniques will be applied to identify appropriate models describing the experimental data obtained in WP 01, and 03. We will select minimal-complexity models that adequately account for the data and make testable experimental predictions. To describe the dynamics of the pathway in different cell types (endothelial cells of the gut versus infiltrating monocytes, macrophages and lymphocytes), we hypothesise that they are caused primarily by different expression levels of receptors (TNFR, TLRs) and pathway components. We will quantify important components experimentally and include this information explicitly into the models to arrive at cell-type specific models of canonical and non-canonical NF-kappa-B pathway dynamics in response to TNF-alpha and microbial products, respectively. This mechanistic insight into how cell-specific NF-kappa-B signalling patterns are generated will lead to testable predictions about how one could selectively interfere with them for therapeutic purposes.
Task 3: Dissecting cell-to-cell heterogeneity
To study tissue-level effects it will be important to consider heterogeneity between cells. Building on deterministic models developed in Tasks 02.1-2, we will account for intrinsic noise by developing stochastic models. Where necessary, we will also include cell variability in expression levels of pathway components. In-house and published stochastic models are available and will be further developed.
Task 4: Modelling target gene activation by NF-kappa-B
Microarray time series show that TNF-induced NF-kappa-B target gene expression varies substantially between different treatments; suggesting that subsets of reporter genes might act as biomarkers. The available data sets will be used to inform modelling of target genes. Moreover, recent work has shown that mammalian gene regulation is intrinsically dynamic involving a complex interplay of protein recruitment and chemical energy-driven enzymatic modifications of chromatin and RNA polymerase. Complex dynamics result in slow timescales for reversible gene activation, implying that dynamic patterns of NF-kappa-B signalling might selectively activate sets of target genes. We will model the interface between NF-kappa-B activation target genes regulations using a stochastic framework. Starting with a generic model for the transitions between inactive, poised and active promoter states as well as initiation, elongation and pausing of RNA polymerase, we will analyse which parameters of the NF-kappa-B signal (NF-kappa-B amplitude in the nucleus versus number and affinity of genomic binding sites; NF-kappa-B oscillation period versus timescale of a gene?s activation-inactivation cycle) determine the response of different sets of genes. We will connect this mechanistic analysis with time-resolved transcriptome studies in WP08, analysing both primary NF-kappa-B target genes and genes driven synergistically by NF-kappa-B and other TLR-activated transcription factors (e.g., IRFs) in conjunction with bioinformatic work (WP04, WP09). This will help rationalise single-cell transcriptome studies and inform the search for new biomarkers for IBD progression.
Task 5: Role of cell-to-cell communication in shape tissue-level NF-kappa-B signaling
Cell communication via cytokines plays a key role in the development of IBD. Inflammation triggered by gut microbes activates NF-kappa-B via TLRs, and the subsequent cytokine release activates and recruits pro-inflammatory immune cells (Th1 and Th17 lymphocytes, macrophages/monocytes). To dissect this dynamic interplay and inform the design of co-culture experiments (WP01, 03), we will model intercellular communication via cytokines. Based on previous work (Busse/Höfer PNAS 2010), we will develop multi-scale reaction-diffusion models that link intracellular signalling with paracrine cell communication to understand tissue-level NF-kappa-B signalling patterns in the gut. In parallel, we will develop computational tools to build, simulate and document multiscale models at the tissue level. Thus we will extend existing standards for representation of large biochemical networks with respect to including their spatio-temporal organisation. The resulting tissue-level models will lead to testable predictions on key factors that determine the transition from spatially localised signalling patterns that affect a limited number of cells to large-scale propagation and synchronisation of signalling.