WP08 - Genomics
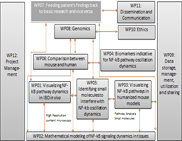
Objectives
The central genomics project will analyze the genomic, transcriptomic and epigenomic data corresponding to the experimental and clinical work packages of the consortium. The project comprises work with different
sample input types (cell line work, patients? mouse models). An in depth characterization of transcriptomal and TF-binding signatures (p50/p65 ChIP-Seq) will be performed according to roadmap guidelines of international
RNA sequencing consortia (ICGC/IHEC). We will further analyze the genomic and, in selected cases, also the epigenetic variability of the employed samples by exome sequencing and/or ultradeep (epi) genotyping
approaches. We aim to provide ultra deep data sets for systems biology modelling of NF-kappa-B dynamics in order to understand:
- the dynamic transcriptional landscape downstream of NF-kappa-B activation from abundant to rare RNAs(non-coding RNAs, alternative mRNA splice isoforms, sense/antisense ratios) (task 1)
- analyze the impact of NF-kappa-B activation and DNA binding on kinetics of transcript processing (splicing, alternative isoforms) (task 1, 2)
- the contribution of allelic variation to individuality and plasticity of NF-kappa-B-dependent transcription (task 3)
- the modulation of small non-coding RNAs including miRNAs by NF-kappa-B signalling and vice versa the impact of miRNAs on NF-kappa-B-dependent events (task 1).
Workpackage Description
The work package performs a primary mapping using different mapping strategies (for transcriptomes: GSNAP; segemehl, TopHat) which will be compared to available genomic sequence information and prepared in a standardized output format for analyses in the Systems Biology work packages using transcriptome analysis packages (currently, Cufflinks, T-ACE, mirDeep). Genome/epigenome information will be analyzed using BWA, GEM, B-SOLANA and various SNP callers, e.g. GATK/SAMtools. The ICMB will maintain a secure site for sequence data download, a system for tracking project management, storing of SOPs for experiments and data analysis.
Task 1: RNA-seq: Purified fractions of small RNAs and full-length polyadenylated mRNA will be subjected to fragment library construction and sequenced using the Illumina Hiseq technology. The protocol preserves the strandednes of the information and thus allows for an estimated sense/antisense ratio at each nucleotide position. We have developed specialized protocols for the analysis of ultra-low input (i.e. nearly single cell) samples or GRO (global run on) seq for studying transcript processing by metabolic labelling. For small RNA after the appropriate fractionation and extraction of RNAs with size ranging between 10 and 40 nt, sequencing will be performed using standard Illumina small RNA sequencing protocol (single end 50nt) with a minimum of 10 million reads for each sample. For the long RNAs (cRNA and ncRNA, polyA), each sample will be sequenced in one lane with 100nt from both ends (Paired End), with a minimum of 100 million sequencing read pairs (month1-60).
Task 2: Chip-Seq: Nuclear chromatin DNA will be sheared and ChIP-seq using anti- p50/p65/modified histone (e.g. H3K9me3, H3K27me3) antibodies will be performed according to established standard protocols (100 M reads/sample (month 1-60).
Task 3: Exome-seq/(epi)genotyping: For selected human samples, it is important to analyze the genetic variability behind the observed dynamics of NF-kappa-B activation. For this, both exome sequencing covering the coding regions of the human genome or high density SNP typing (Illumina iSCAN) is available. Methylation as a stable epigenetic mark will be investigated using RRBS-seq or 450k Illumina Infinium assays. (Month 1-48)